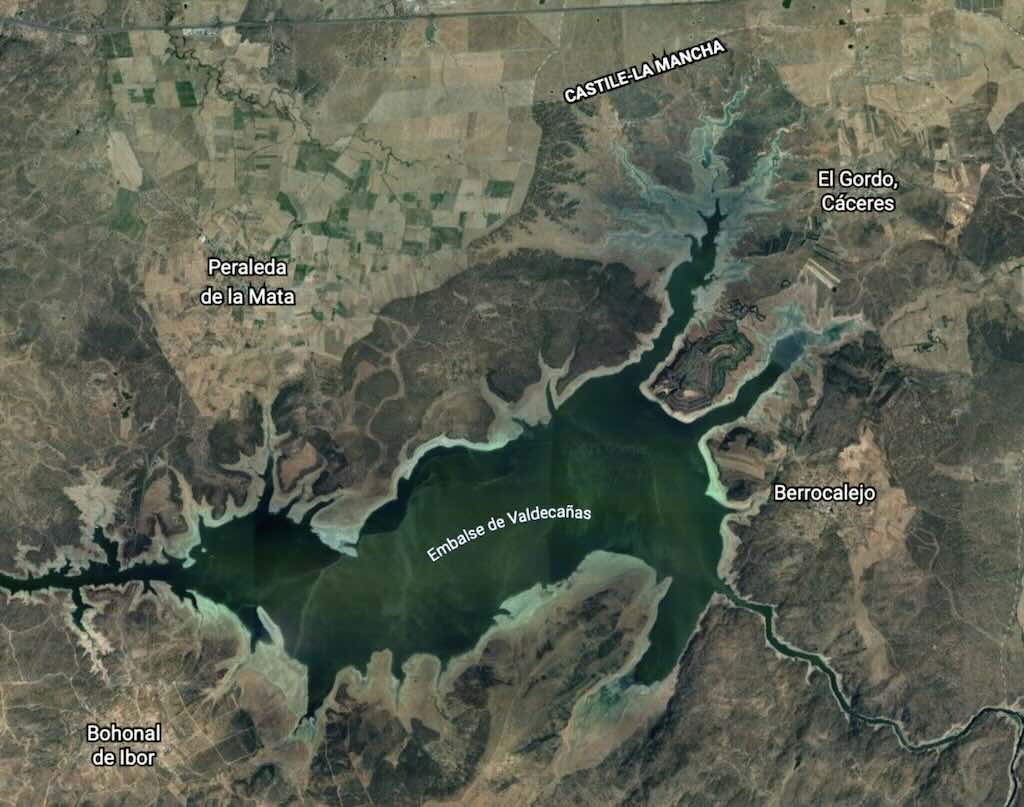
One of the big evolutions in the uranium program after June 1940 was that isotope separation became increasingly important. As information became available about the fission of uranium-235 by slow neutrons, the need to consider ways to separate uranium-235 from uranium-238 emerged as a new priority. Contracts for the development of gaseous-diffusion and gas centrifuge separation processes were recommended by the Planning Board, which would also be responsible for the heavy-water production program.
On October 21, 1941 Arthur Compton produced a status report on isotope separation. The results of the MAUD committee were reviewed, Robert Sanderson Mulliken reported on his visit to the different research teams, and Julius Robert Oppenheimer suggested that about 100 kg of uranium-235 would be needed for an effective weapon. I think this is the first time we see the mention of Oppenheimer in an official meeting. At the time he was a young physicist in Lawrence’s team at Berkley. Broadly speaking Lawrence did not like the way meetings tended to focus on uncertainties, and not on the possibilities. The report issued on November 6, 1941 concluded that a “fission bomb of superlatively destructive power” was possible if they had sufficient uranium-235. ‘Sufficient’ was between 2 kg and 100 kg. I think for the first time they also mentioned that the bomb would be fired by bringing together parts, each less than a critical mass. So speed of assembly would be essential. Do it wrong and it would just ‘fizzle‘.
The consensus was that gaseous-diffusion looked feasible, but might take time to develop. The centrifuge method appeared practical and was more advanced than gaseous-diffusion. Other alternatives, such as using a mass spectrometer, were still in the laboratory phase. The time scale was three to four years, and at a cost of $50 to $100 million. The suggestion was that trial units of the centrifuge and gaseous-diffusion systems should be built and tested. The conclusion was that engineering was becoming the priority. What was not mentioned was the use of element-94 as a substitute for uranium-235. Yet Compton was of the opinion that the chain reaction would lead to a valuable power source and to the production of element-94 which could become another practical means to produce a fission bomb. This alternative was dropped in order to focus attention on a military result that could be useful in the current war. In any case the Fermi ‘pile’ was still not a given, so there was no way that large quantities of element-94 could be produced.
Bush sent the report of November 27, 1941 to Roosevelt with a covering letter saying he intended to form an engineering group and accelerate the physics research aimed at plant-design data.
In the re-organisation following Pearl Harbor task assignments were better defined. The Planning Board was responsible for engineering, and that meant pilot-plant experimentation and any large-scale laboratory investigations. Urey was responsible for isotope separation methods (diffusion and centrifuge) and heavy-water investigations. Lawrence was responsible for small-sample preparation, electro-magnetic separation, and ‘certain cyclotron work’ (meaning element-94). Compton was responsible for atomic physics, including weapon theory and chain reaction (including element-94). It was at this time that Vannevar Bush stated that when full-scale contribution started, the Army would take over.
Naturally when writing about isotope separation it is perhaps useful to first look at isotopes, and in particular the work done in 1940. Ernest Orlando Lawrence had received the 1939 Nobel Prize in Physics for the invention of the cyclotron. He was director of the Radiation Laboratory at the University of California, and in spring 1940, with Edwin Mattison McMillan and Philip Abelson, discovered element-93, and McMillan suggested the name neptunium. It was in December 1940 that Glenn Theodore Seaborg with Joseph William Kennedy with Arthur Charles Wahl finally confirmed the idea of McMillan and found evidence of element-94 (it was chemically identified in February 1941). The key was the existence of the 60-inch cyclotron. They were able to bombard uranium with deuterons and produce samples of significant size. The actual discovery was only announced after the end of WW II, but numerous scientists, including Bohr and Wheeler, had predicted the existence of transuranium elements. Initially element-94 was just seen as another component of the atomic ‘pile’, but it quickly became evident that if it were produced in a ‘pile’ and chemically separated they would have an alternative to uranium-235. Even better, they may not need to build expensive isotope-separation plants. It was Seaborg’s team and Emilio Gino Segré who bombarding uranium with neutrons were able in May 1941 to confirm that element-94 would fission with slow neutrons. By the end of the month it was known that it was about 1.7 times as likely as uranium-235 to fission with slow neutrons.
It was about the same time that Lawrence decided to convert his ‘old’ 37-inch cyclotron into a big mass spectrometer, paving the way for the electro-magnetic isotope separation.
The scientific aspects of the work on isotope separation was kept apart from the procurement and engineering phases. The Program Chiefs H. C. Urey, E. O. Lawrence, and A. H. Compton were to have charge of the scientific aspects. Initially it was proposed that Urey should have charge of the separation of isotopes by the diffusion and the centrifuge methods and of the research work on the production of heavy-water. Lawrence was to have charge of the initial production of small samples of fissionable elements, of quantity production by electro-magnetic separation methods, and of certain experimental work relating to the properties of the plutonium nucleus. Compton was to have charge of fundamental physical studies of the chain reaction and the measurement of nuclear properties with especial reference to the explosive chain reaction. He was authorised to explore also the possibility that plutonium might be produced in useful amounts by the controlled chain reaction method.
The effect of the reorganisation was to put the direction of the projects in the hands of a small group consisting of Bush, Conant, Briggs, Compton, Urey, Lawrence, and Murphree. Theoretically, Compton, Lawrence, Urey, and Murphree were responsible only for their respective divisions of the program.
The cost of the initial work was estimated at $4-5 million, and it was stated that the Army would take over when full-scale construction was started, presumably when pilot plants were ready.
In a meeting of the reorganised S-l Section Conant called for an all-out effort given the military value of atomic bombs and that all attention must be concentrated in the direction of bomb development.
The Technologies of Isotope Separation
Isotopes differ only in the number of neutrons in the nucleus, but not in atomic number. For this reason separation of isotopes is usually based upon the mass difference and not on a chemical method. For the scientific community the centrifuge appeared to be the best option. The principles were simple, even if there were technical problems in spinning the separator fast enough. No one thought these difficulties were too serious. Purely mechanical problems were expected to yield quickly to a concerted research effort.
Gas Centrifuge
So we will start with the gas centrifuge. Today this technology is seen as small, inexpensive, and relatively simple to make. It is the device that is best known for making nuclear weapons available to the developing world. It was originally developed and tested for separating two chlorine isotopes, and through to 1944 it was one of the isotope separation technologies developed in the Manhattan Project. However the research of Harold Clayton Urey and Karl Paley Cohen at Columbia University was discontinued because it felt that it would not produce results before the end of the war. After WW II it was German prisoners of war who helped perfect the centrifuge in a Soviet labour camp. In fact the centrifuge was never a sophisticated or resource-intensive technology, and we now know that it was in fact technical feasible during the Manhattan Project, but it was not successfully developed. The project was shut down before the developers had the opportunity to solve some of the technical shortcomings.
Soviet spies quickly learned about those shortcomings and when the German team finally did solve the problems with the centrifuge it turned out to be an incredible simple device in which no precision engineering was required. It was for this reason that the Chinese started a centrifuge program as early as 1957 (a program that took decades for the U.S. to detect). By 1975 India, Brazil and Pakistan all had centrifuge programs, and Pakistan passed on the technology to North Korea in the mid-1990’s. Today experts have estimated that at least 20 countries have built or acquired centrifuge technology.
The existence of isotopes were first announced in 1918 by Frederick Soddy and John Arnold Cranston, and it was this work that inspired H.G. Wells to write about atomic bombs being dropped from biplanes in The World Set Free. It quickly became evident that the only way to separate chemically identical atoms of the same element was to exploit their minute difference in masses and their radioactive properties. It was Frederick Alexander Lindemann and Francis William Aston (the inventor of the mass spectrometer) who wrote at the time that the centrifuge was the most promising method for isotopes separation. However throughout the 1920’s the engineering problems proved formidable. The simple problem was that no one could get the centrifuge to spin fast enough. Finally when they could spin it fast enough the air friction heated the centrifuge so much that it cancelled the separative effect. It was Jesse Wakefield Beams in 1934 who had the insight to build a centrifuge inside a vacuum chamber, and he managed to separate chlorine-35 from chlorine-37 (the two stable isotopies of chlorine).
In fact the early centrifuges were based on a shaft-mounted rotor, and were known from early times but it was Karl Gustaf Patrik de Laval in 1883 who introduced the flexible shaft that was able to shift under the force. It was Theodor Svedberg, a colloid chemist, who in the 1920’s invented the first ultracentrifuge, machines that work at 1,000’s rps. His centrifuges were housed in a low-pressure hydrogen atmosphere to reduce friction and equalise temperature differences. His research earned him the 1926 Nobel Prize in Chemistry for providing evidence of the physical existence of molecules. Émile Henriot investigated ways to spin tops at extremely high angular velocities using air-jets, and the technique was used to construct the shaft-less ultra-centrifuge (Henriot was also the first person to show that potassium and rubidium are naturally radioactive). Examples of these shaft-less centrifuges could spin at 4,000 rps, however the next limitation was friction with the atmosphere. Beams placed his rotor in a high vacuum. His first centrifuge used a flexible shaft attached to an air-drive turbine and suspended in a vacuum. As with earlier systems this design with a flexible shaft allowed the rotor to spin about its own inertial axis. The next step was to try to use magnetic fields to support the rotor. Beams design used an external electro-magnet to lift the rotor and let it spin freely and rotate about its own inertial axis. The problem with this design was that the rotor could move up and down if the balance between the magnetic field and gravitational pull was not maintained. Beams used a beam of light and a photoelectric cell to obtain positional information and feed that to the current powering the electro-magnet. This design could spin at 1 million rps. One of Beams students would go on to design a field-supported centrifuge that would be commercialised as a sample preparation tool found in almost every laboratory (this ultracentrifuge was marketed by the industry leader Spinco). Beckman bought Spinco in 1955, and the ultracentrifuge was used to prove James Watson and Francis Crick‘s theory on DNA replication. Through into the 1970’s the analytical ultracentrifuge was one of the most conspicuous and valuable tools in microbiology and biochemistry laboratories. There is a world of different between a very small object spinning at 1 million rps for short periods of time, and a laboratory ultracentrifuge from the 1950’s that was the size of a small car and could manage up to 100,000 rpm.
Already from 1940 the centrifuge was the preferred method for the large-scale enrichment of uranium. Beams and Alfred Otto Carl Nier had separated small uranium samples in a mass spectrometer and neither felt that the technique was scalable. On the other hand Beams was convinced that the centrifuge was potentially scalable for uranium enrichment. From 1941 the largest share of funding went to Beams and the centrifuge, four times more than that allocated for gaseous-diffusion. Initially a small centrifuge was designed at Columbia University and built by Westinghouse Research Laboratories, and a ‘high-performance model’ was developed by Beams at the University of Virginia. These machines were based upon the laboratory apparatus that was used to separate chlorine isotopes. The approach of Beams was to use the funding to test a primitive single-batch centrifuge concept called the ‘evaporative centrifuge’ on uranium hexafluoride (UF6). Most experts now consider this as a poor use of wartime funding. In fact the engineering development had already started with Westinghouse before the first meeting of the Planning Board in July 1941.
We start with a binary gas mixture of two molecules, one slightly lighter than the other. In very simple terms the gas enters the centrifuge and undergoes a centripetal acceleration. The rotor is turning at a constant speed (constant tangential speed), but the gas molecules will feel an acceleration (change of direction of the velocity) as they move in the uniform circular motion, and that acceleration is in the direction of the change of velocity, which is pointing towards the centre of rotation. But the molecules feel an apparent force, equal and opposite to the centripetal force (the centrifugal force), drawing them away from the centre of rotation, and this is caused by the inertia of the molecules. So the denser or heavier molecules move towards the exterior wall of the rotor, and the lighter molecules don’t move as much and remain closer to the centre. The lighter fraction is richer in uranium-235 as compared to the heavier gas fraction which has been depleted of uranium-235 and is thus heavier with uranium-238. In any one centrifuge the different is small, so in order to be effective several centrifuges are connected in series (a cascade).
One of the key features is the way the centrifuge changes the direction of enrichment from radial to axial, by inducing a weak axial circulation of the gas, called a counter-current flow. It is this ‘trick’ that allows the centrifuge to remove enriched and depleted gases at each end. What it creates are two streams going in opposite directions from one end to the other. The stream nearer the axis is becoming enriched, whereas the stream near the outer wall is becoming depleted. In this sense the centrifuge acts as a kind of distillation column. The reason for creating a counter-current flow is the axial separation factor is much larger that the radial separation factor. This counter-current can be created by heating the bottom end and cooling the top end. The other method is mechanical, where a stationary object (disk) is placed in the gas stream near the bottom end. The scoops can act as mechanical obstacles as well, creating a drag which changes the tangential velocity of the gas and at the same time warms the gas. These two effects can also create the counter-current.
The overall performance of a centrifuge is related to the shape of the flow profile and the strength of the counter-current. In theory the performance has a 4th-power dependence on the peripheral speed of the rotor and is directly proportional to the length. We can see from this the importance of the rotor speed.
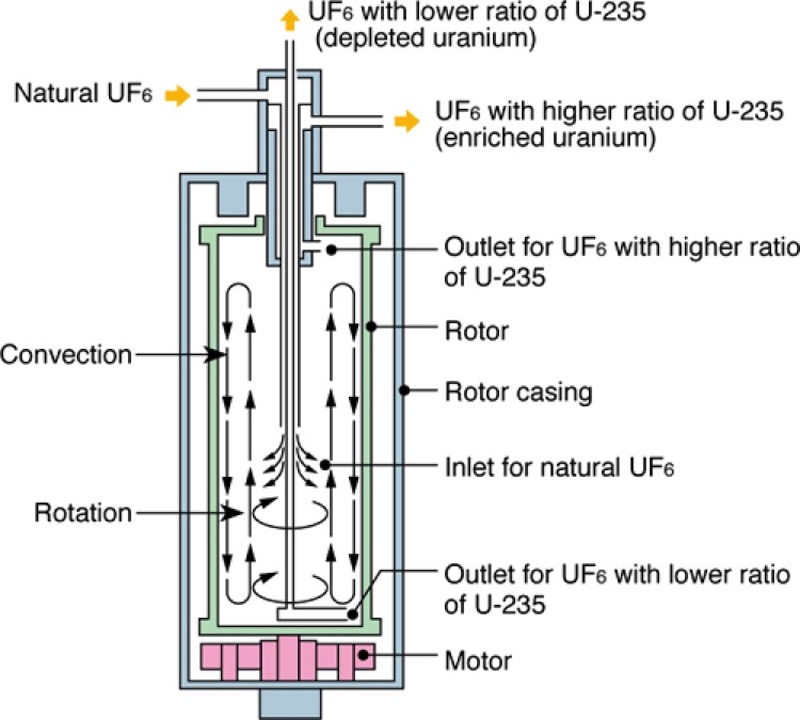
What we can see above is the so-called short bowl centrifuge developed after WW II (see below for further details). We can see some of the basic features. We can see the rotor in green, which spins around its axis sitting on a thin, flexible steel needle bearing (today the speed is about 22,000 rpm or about 350 m/s for smaller centrifuge units). The idea of the needle bearing is that it is not a rigid bearing but it allows the rotor to pivot like a top. The needle is sitting in a depression in a hard metallic plate, which in turn is centred elastically and is damped by oil friction. The upper bearing of the rotor consists of a hollow cylindrical permanent magnet mounted in a damped gimbal through which a steel tube passes without touching the wall. Three tubes for the insertion and extraction of the gas pass through this opening. You have a feed tube which ends in the middle of the rotor. The other two exit tubes actually terminate at opposite ends of the rotor (not easy to see in the diagram above but they are marked ‘outlet’). These tubes have their ends bent into a hook-shape curve (‘scoops’) perpendicular to the axis of rotation. These scoops make use of the high impact pressure in their vicinity to transport (extract) the heavier and lighter gas fractions. The extractor tube for the enriched uranium hexafluoride has a slightly reduced angular velocity (as compared to the other tube) which induces the counter-current flow. The extraction tube for depleted uranium hexafluoride is in fact protected by a lower plate or baffle which is not shown in this diagram. This baffle has holes in it to allow the gas to leak from the main rotor cavity into the area near the scoop. The baffle is needed at one end to stop the scoop imposing a vertical flow that would counteract the circulatory flow generated by the scoop at the other end. The vacuum cylinder or stator (the blue part near the top nozzle) has screw type groves in the wall, and combined with the rotating tube act as a molecular pump to maintain a vacuum around the end of rotor, reducing gas friction, and thus any heating. We can see the electric motor where the armature is a flat hardened steel plate attached rigidly to the bottom of the rotor. The field winding (which is marked motor) is located in the vacuum and is fed from outside by an alternator at a frequency synchronous with the speed of the rotor.
Wikipedia has an interesting article about both Gernot Zippe and Manfred von Ardenne. Zippe had worked on isotope separation for the German nuclear weapons program. At the end of the war he was kidnapped and imprisoned in the Soviet Union. He was forced to work on a centrifuge project in a physics institute near Sukhumi, a small seaside resort town in Georgia (the institutes were often called sanatoriums). The institute was run by Manfred von Ardenne who after WW II agreed to work for the Soviet Union on isotope separation as part of the Soviet atomic bomb project. Although the centrifuge option was abandoned during the Manhattan Project the Soviet Union continued to develop the technology. Through 1947 the objective was to study mechanical and metallurgical problems associated with the rotor speed. They were able to attain about 80,000 revolutions/minute (rpm), but the target was 150,000 rpm. The next step was using a ‘special aluminium alloy’ for a reinforced rotor 40 cm long. A considerable number of tests were made to eliminate vibration and any unbalanced movement. Sleeve and needle roller bearings were used to reduce friction, and all the bearings were cooled in chambers of liquid air. In particular a needle bearing allowed the centrifuge rotor to spin on a needle point, with very little friction. This type of ‘point’ bearing was an adaption of the jewel bearing used in watchmaking and in some electric motors dating from the 1900. The other types of bearings were made a bit more flexible and with weak damping, allowing the centrifuge to adjust itself so that it could spin on its axis without vibration. Using tight bearings and strong damping often forced the centrifuge to spin off-axis and vibrate. This idea was an implementation of Karl Gustaf Patrik de Laval‘s principle of self-balancing, used for steam turbines since 1889. In addition the internal walls of the rotor were lined with permanent magnets, and the rotor itself was driven asynchronously by a high frequency transmitted via a continuous winding wrapped around the rotor chamber. The idea of an indirect drive came from an indiction-welding system build by another German POW working in the same institute. With all these innovation they finally attained 150,000 rpm. The key appeared to be proper cooling using both air and water. An efficiency claim for just one unit was 2%. Zippe has a specific type of centrifuge named after him, the Zippe-type centrifuge. He finally was released in 1956 and for a period worked with Beams at the University of Virginia, before retiring back in Europe. The work Zippe did in the University of Virginia concerned what he called the short-bowl centrifuge, and the reports are in the public domain (the photograph below is the prototype built at that time).

The experts who have looked back over the development of the centrifuge in Russia noted that already in 1940-41 they too were interested in thermal diffusion and the gas centrifuge. They too felt that the thermal diffusion had the greater initial research base, but many felt that it would consume more power than could be extracted from the ‘purified’ uranium-235. The only centrifuge program at the time was run by a German émigré called Fritz Lange. His centrifuges were heavy, noisy, and clumsy machines that operated horizontally on roller bearings and produced no measurable enrichment. The Russians at the time thought that the U.S. was building a nuclear bomb, so they also began to explore that possibility. Georgy Nikolayevich Flyorov also worked out that a fast-fission chain reaction was necessary and that enriched uranium would be required. It would appear that the Russians were also aware of the MAUD report, so the laboratory of Igor Vasilyevich Kurchatov also took on the work of Lange on centrifuges. The initial design of Lange was about 60 cm long and 25 cm in diameter. Internally the rotor was divided into 60 chambers, and the total weighed around 2½ ton and consumed 3 kW. The key to its failure was that the rotor’s maximum speed was only 130 m/s, well below the necessary speed to effect measurable separation. Lange tried to improve on the design by making the centrifuge longer, but what happened is that the 5 m long rotor sagged in the middle and caused destructive vibrations (and at high speeds they could snap in the middle). The bearings on the Lange centrifuge only lasted eight to ten hours, and not the 99 days with Beams’s centrifuge. In 1944 Kurchatov learned that the U.S. had chosen diffusion over centrifuges, and he was receiving information from the spy Klaus Fuchs who was working on diffusion membranes. It was at this time that Lange was transferred away from the laboratory as it turned to gaseous-diffusion. This was probably a mistake because the centrifuge ultimately proved the more efficient, however a horizontal centrifuge would never have worked (Beams had published some 6 years earlier that they were using vertical centrifuges). It was the German Max Christian Theodor Steenbeck who offered to work for the Russians on centrifuges and he started with copying the work of Beams. Gernot Zippe worked in that team.
In 1949 the Russians discovered, like the Americans, that with gaseous-diffusion they could not get past 40% enrichment. This gave Steenbeck the chance to propose to use centrifuges for a top-up plant to go from 40% to 90%, but it was not certain that the design was ready for mass production. The centrifuges were expensive to build and operate. At the time they relied on external vacuum pumps to maintain the vacuum seal around the rotor, and in addition they needed compressors to pump the uranium gas from one centrifuge to another. An industrial application would consume enormous amounts of electricity, as did gaseous-diffusion plants. Steenbeck proposed, as did Beams, to build 5 m long centrifuges, reducing the number of vacuum pumps and compressors needed. A new problem occurred, the long rotor was not rigid enough and vibrated. The solution was to use shorter rotors interconnected by bellows to compensate for the vibrations. However, a certain Voskoboynik solved the problem with gaseous-diffusion plant, so the top-up option was never needed. Work continued to 1952 where the entire operation was transferred to Leningrad and integrated into the Soviet nuclear production program. It was there that the engineers added a pitot tube to extract the gas from the centrifuge. The tube harnesses the rotational momentum of the gas to pump it from one machine to the next (an idea that had been used in centrifuges since 1901 to eliminate compressors). They also added a spiral-grooved sleeve around the top of the rotor so the outside of the spinning centrifuge tube acts as a self-evacuating vacuum pump, and idea called a Holweck molecular pump invented in 1922. All the improvements meant that there was no need to move to long centrifuges, and they focussed on building short, simple centrifuges. A plant came online in 1957 and had 2,435 centrifuges by 1958. Intelligence estimates put the production of such a plant at one highly-enriched implosion device every two years. According to Zippe’s estimates 20,000 centrifuges of this type could produce about 1 kg of 96% enriched uranium per day. This assumes 100% efficiency, but in practice 20% efficiency was a feasible target.
Today the reality is that the gas centrifuge is the most economically efficient way to enrich uranium for peaceful power-reactor fuel, but a so-called ‘rapid-breakout’ would also enable a country, using the same technology, to produce nuclear weapons before there is time for a political response. In addition clandestine plants are difficult to detect as compared to nuclear reactors or large gaseous-diffusion plants. Centrifuge plants use little electricity and produce little detectable signal, so the plants are much easier to hide. Check out this article for an extended review of centrifuge technology.
At a meeting on November 10, 1942 it was decided to pursue a full-scale gaseous-diffusion plant, with the electro-magnetic method held in reserve, and to drop the centrifuge. These decisions were taken after an extensive review that showed that, despite initial high expectations, the centrifuge had not met those expectations. Many reports on the Manhattan Project don’t mention the centrifuge after that date, however other reports indicate that the centrifuge enjoyed a brief revival a few months later.
Gaseous-Diffusion
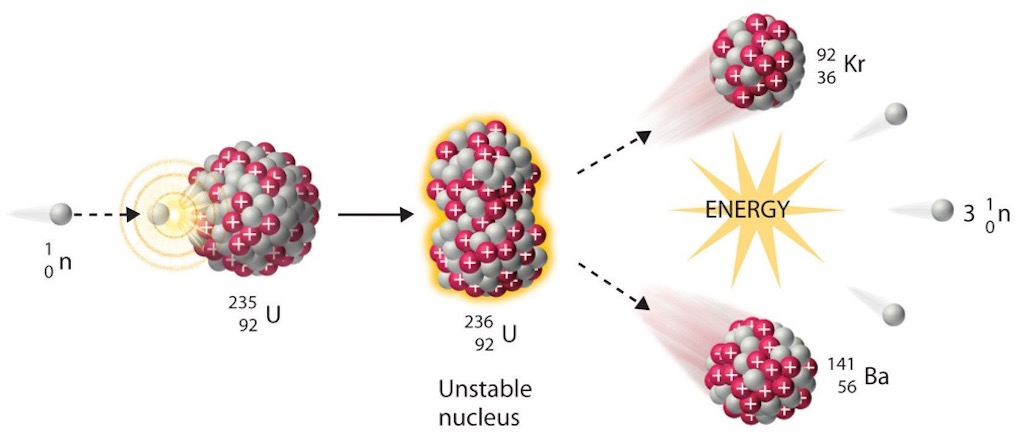
After nuclear fission was first discovered, the next step was the determination that the fission reaction was due to the isotope uranium-235. Once the idea that a self-sustaining chain reaction might be possible, the problem became one of trying to concentrate uranium-235 relative to the far more abundant uranium-238. As mentioned elsewhere the task was to use the difference in atomic masses between the two isotopes, and the difficulty was that the difference is very small.
The ‘classical’ method to separate two gases is gaseous-diffusion, based upon the experimental finding of Thomas Graham in 1829 who pioneered the study of the diffusion of gases. He found that the rate of effusionof gases through small apertures was inversely proportional to their molecular weights. Francis William Astonhad used the principle of Graham to partially separate the neon isotopes. Later William Draper Harkins would apply the method to chlorine, and Gustav Ludwig Hertz achieved almost complete separation of neon isotopes, by recycling the gas through many stages.
The basic physical underlying the gaseous diffusion method is the so-called ‘equipartition principle‘ of statistical mechanics. The basis is that in a gas consisting of several types of molecules each type will have the same average energy of motion (kinetic energy). This equality of average energies is attained and preserved by the enormous number of collisions between the molecules in the gas. These collisions ensure that any excess energy will be rapidly shared equally with all the molecules, this is the meaning of ‘thermal equilibrium‘. The kinetic energy of a molecule is related to its mass and it’s velocity, therefore molecules having the sane kinetic energy will have average velocities which differ in inverse proportion to the square roots of their masses. If we take a uranium hexafluoride molecule with uranium-235 and another with uranium-238 the ratio of their velocities is 1.0043 with the lighter molecule moving very slightly faster ‘on average’. The idea of gaseous-diffusion is to exploit this small different in average velocities by forcing the gas mixture to diffuse through a porous barrier under a pressure difference. The faster molecules will encounter the holes more often than the slower ones, and therefore be slightly more likely to pass through the barrier. The gas that emerges on the other side of the barrier will be slightly ‘enriched’ with the lighter isotope. The ideal of 1.0043 is never met since it’s dependent upon the concentration of the desired isotope on the ‘feed’ side which will gradually decrease as the enriched mixture diffuses through the barrier (a value of 1.0014 would be good practice).
A uranium hexafluoride molecule has a diameter of about 0.7 nm and at a pressure of about ½ atmosphere and at a temperature of about 80°C the average separation between molecules will be about 5 nm. The mean free path under these conditions is about 85 nm, so the average pore size in the barrier must be somewhat less, say 25 nm. To get this pore size you could imagine packing spheres together of about 100 nm, which would leave gaps of about 25 nm. So one could imagine a barrier made of sintered nickel power, i.e. packed under high pressure and at a high temperature. And naturally this barrier must be very thin (few micro-meters) yet withstand a pressure differential of 0.2 to 0.5 kg/cm2. In most cases the barrier would be prepared as a tube made of the thin porous layer deposited on top of a structural layer allowing the barrier to work under higher pressures.
It was on May 21, 1940 that George Bogdanovich Kistiakowsky suggested gaseous-diffusion as a possible way to separate uranium isotopes. At the time he was thinking of the work of a certain Charles G. Maier in developing ways to separate mixed gases. Later he found that the approach of Maier had some important defects, but that the technique looked appealing. It was in early July 1940 that Urey suggested a barrier made of special glass, and initial reports by Kistiakowsky on October 14, 1940 looked promising. He would bow out of the topic due to work pressure, but Urey, Dunning, and others in Columbia University continued through to the spring 1941 to look at alternative barrier materials. The problems were considerable. The barrier needed to have billions of holes with a diameter of about 1/10,000th of a millimetre. Yet the barrier needed to withstand a considerable pressure difference and mechanical strain, not to mention the highly corrosive nature of uranium hexafluoride. And the process must not leak to the air, otherwise uranium oxyfluoride would be formed and clog the barrier. And industrial plant would need 10,000’s of square meters of the barrier and 1,000’s of stages, and would need to work as a process (not by batch). Yet to the experts the technical difficulties appeared less than with the centrifuge.
There are several different separation processes, the liquid thermal diffusion process being the most significant. Thermal diffusion processes using gases also appeared attractive, however after some test experiments the process appeared impractical for large-scale separation. One lone voice, Philip Hauge Abelson, argued for liquid thermal diffusion, and he received funding to continue that work at the Naval Research Laboratory.
Input from the British (MAUD Committee) suggested that isotope separation using the gaseous-diffusion method offered the best chance of success.
Diffusion and effusion are two slightly different concepts, diffusion is the net movement of atoms or molecules from one region of higher concentration to a region of lower concentration. It is driven by the gradient in chemical potential of the diffusing species, i.e. the energy adsorbed or released due to a change of particle numbers of the species, it’s what also drives chemical reactions and phase transitions. Effusion is the process in which a gas escapes from a container through a hole of diameter considerably smaller that the mean free path of the atoms or molecules. Typically this is a gas leaking through a pinhole, where the leak is driven by a pressure difference between the inside of the container and the exterior.
The approach was to look for a volatile species of uranium, i.e. uranium hexafluoride, as a working substance. The technique consists of a series or ‘cascade’ of diffusion stages. The ‘converter’ in each stage consists of a cooler and a ‘diffuser’. The diffuser is a tank-like piece of equipment which enclosed a ‘barrier’. The barrier is a thin, porous, metallic membrane containing billions of submicroscopic holes.
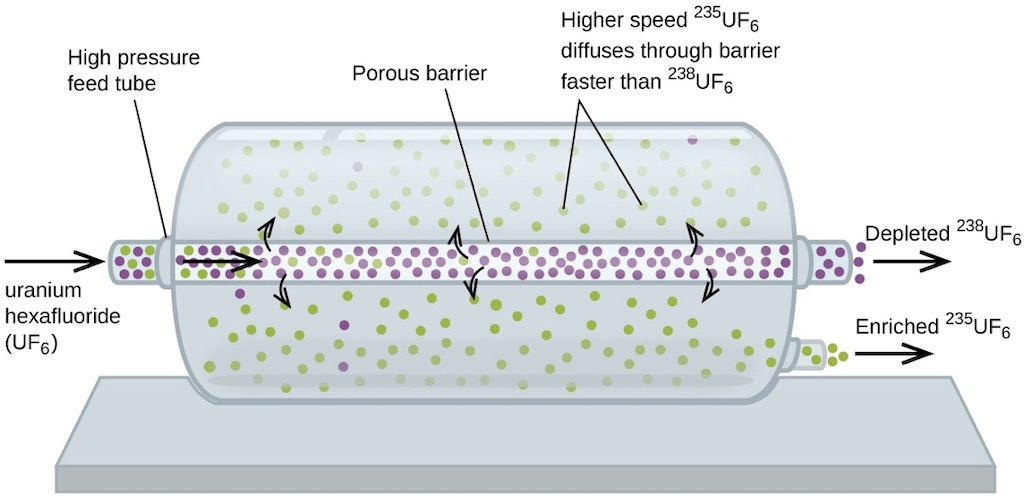
The idea of a cascade is that a pump supplies a converter with a continuous stream of un-diffused gas from the stage above, mixed with diffused gas from the stage below. The cooler is there to remove the heat from the pump and to maintain a constant operating temperature (the cooler removes about 95% of the energy added by the compressor). The idea is that half of the gas fed by the compressor to the converter diffuses through a barrier, becoming slightly ‘enriched’ with the lighter component in accordance with Graham’s Law (i.e. the rate of effusion of a gas is inversely proportional to the square root of the mass of its particles). What this means is that the molecules with a lower molecular weight (uranium-235) have a higher molecular velocity and diffuse more readily through the barrier pores. Consequently, the fraction of the gas that passes through is slightly enriched in uranium-235 and the gas that does not pass though is slightly depleted in uranium-235. The theoretical enrichment of one converter is about 1.0043. The ‘concentrated’ or enriched gas is then pumped to a next higher stage. The remaining half of the gas leaves the converter and is pumped to a lower stage. Naturally the product is withdrawn from the top of the cascade, and the waste or depleted gas is removed from the bottom of the cascade.
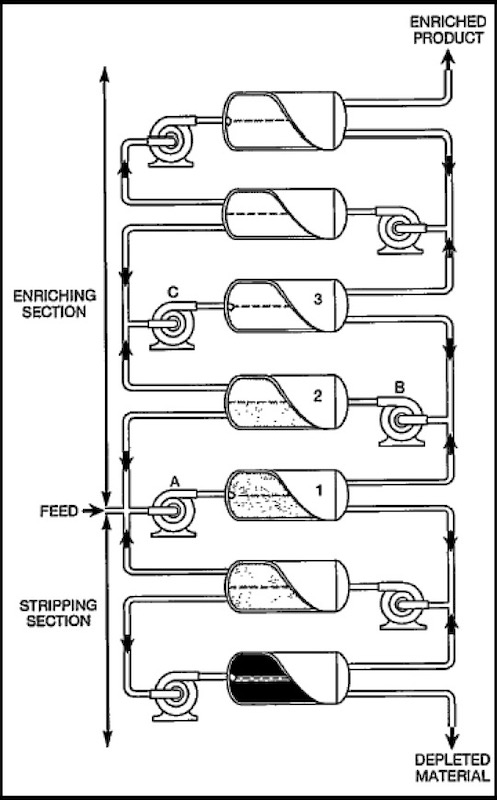
Above we see a schematic of the cascade process, and below a slightly more detailed view of the way converters are linked together in the cascade.
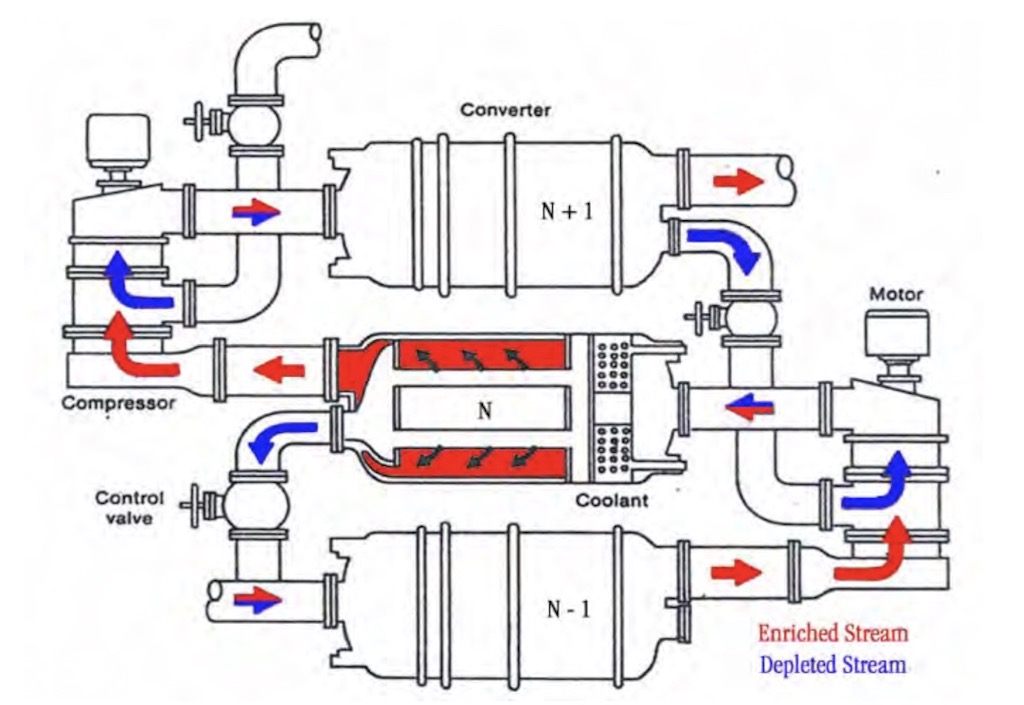
The converter with its associated pumps and coolant, etc. is called a ‘stage’, and today one of these stages is about 4 m in diameter and about 7.3 m long, and contains tightly packed diffusion barrier tubes. Below we can see one stage (note the ‘hard-hat’ in the lower left corner). This is a large converter in the low enrichment section of the cascades, smaller converters are found in the higher enrichment sections.

Several converters are grouped together in a cell, as seen below. A large facility will have 1,000’s of these converters. A large-scale plant could have 5 different sizes of converters, so as to optimise energy consumption, which is in any case significant. Above we see the largest converter which is attached to a 3,300 horsepower motor, whereas the smallest converter might be attached to a motor of only 15 horsepower.
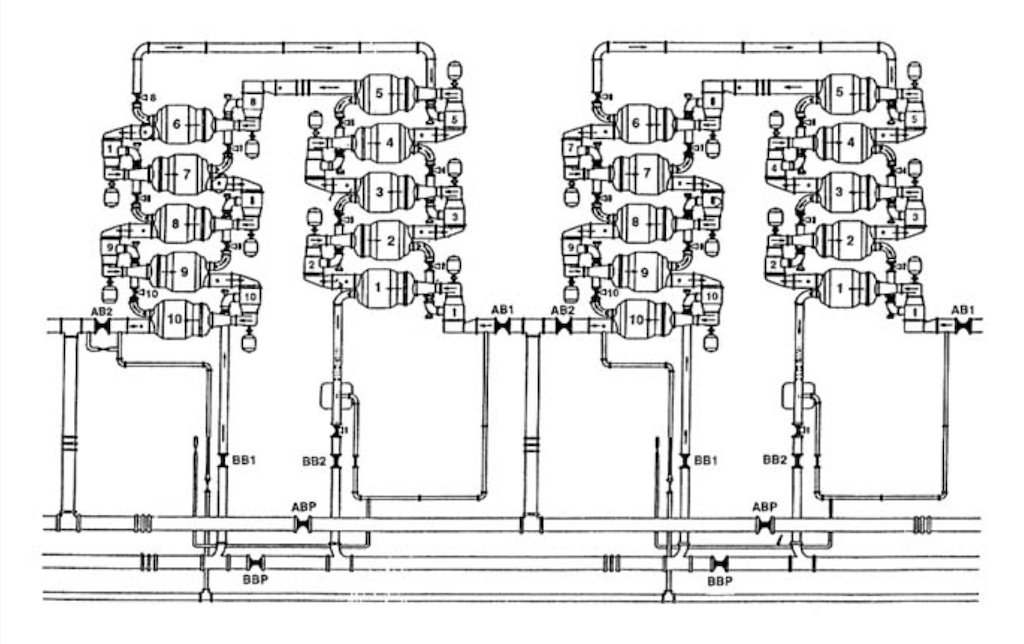

If we look more closely at the barrier, in order for a maximum separation the porous barrier material must meet exacting standards for ‘diffusive’ flow to occur. The pore sizes must be such that individual molecules collide only with the pore walls rather than with each other (see below). In our case to ensure diffusive flow, a uniform pore size of less than two-millionths of an inch diameter must be obtained. The hexafluoride molecule of uranium-235 has slightly less mass than the hexafluoride molecule of uranium-238, thus it can pass through a properly constructed barrier more quickly.
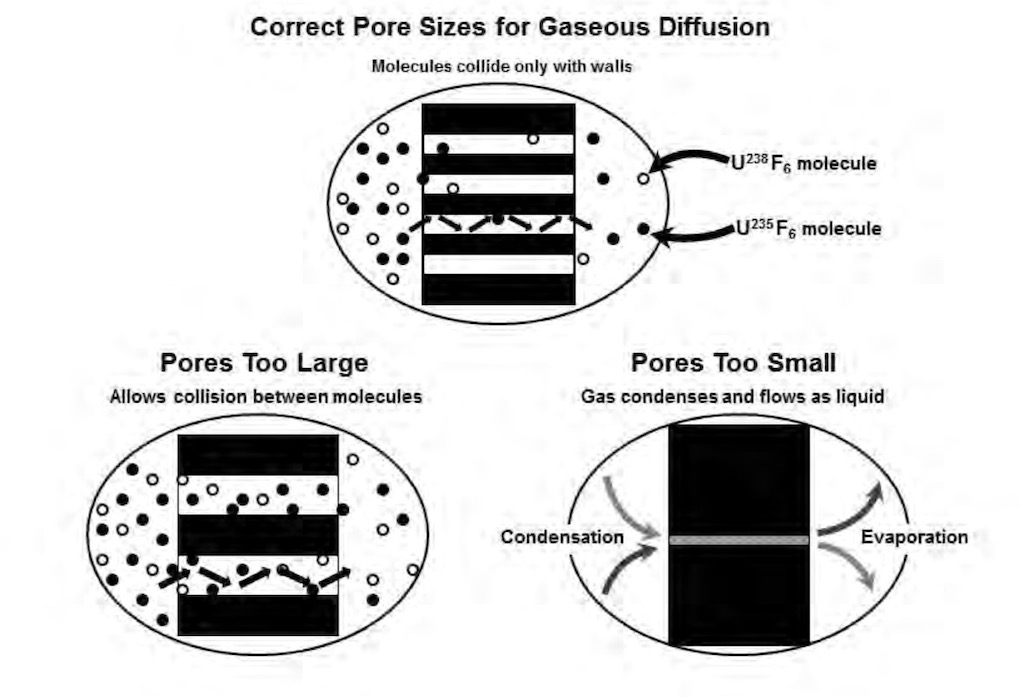
Since the process itself is so simple, there must be some serious technical difficulties. These start with the fact that uranium hexafluoride is extremely reactive with water, it is also very corrosive to most metals, and it is not compatible with organics such as lubricating oils. These system are almost entirely built out of nickel-plated steel, Monel (nickel alloys), and aluminium. The environment in which the uranium hexafluoride is found makes it essential that the barrier retains its separative capability over a long period of time, i.e. the last thing you need is to constantly intervene in such an aggressive environment. Another problem is that because the natural enrichment is very low in uranium-235, the feed will be very large quantities of hexafluoride gas which needs to be pumped through the initial converters.
In 1940 and 1941 the investigation of gaseous-diffusion was limited to testing naturally porous substances, various metal alloys and some metal powers. In addition some tests were performed on the corrosion resistance of some materials, but the laboratory in Columbia University did not even have a pump that could operate with corrosive gases.
When it was decided to build a pilot plant, the first step turned out to be the need to develop components that could withstand the extreme operating conditions. For example there was a need for high-speed pumps that could operate with corrosive gases for extended periods of time without leaks. Another problem was seals and lubricants, and of course there was the ever present problem of the barrier. And not just a barrier, but one that could be produced in 10,000’s of square metres.
Finding the right barrier was the most persistent problem. Francis Goddard Slack tested hundreds of materials, unfortunately certain materials that might work well in the laboratory could not be used in a large-scale production plant. Not only did the material have to undergo tests as barriers, but they also had to be tested for resistance to corrosion. Even the best barriers found during the summer of 1941 were simply (a) not sufficiently corrosion resistant, (b) too brittle and fragile, and (c) were not sufficiently uniform. Attention turned to nickel barriers, which were at least resistant to corrosion.
Lacking information on the barrier, it was difficult to design the converter to be used in the large-scale plant. But next to the barrier the pumps were the biggest problem. The specification was simple, the pumps needed to pump a relatively heavy, highly corrosive gas at high velocities, with no leakage into or out of the system. By summer 1942 they were able to test some different type of pumps with seals etc., but no one was able to produce anything more than a test-tube of uranium-235.
Reading through the reports one is struck by the continued search for a decent barrier by a team of 80-odd people at Columbia University, alongside the work of the Army (with Kellex and Union Carbide) who were trying to specify, design and build a large-scale production plant at Oak Ridge.
Advances were made, they designed and successfully tested a new seal on the pumps, but the barrier remained elusive. After a year of intense research and testing they still had not found a good barrier. By then they would have accepted a ‘bad’ barrier, i.e. one that had stable (but bad) separative qualities, but worked over long periods of continuous operation. How to make sub-microscopic holes that did not clog or plug? How to make a fine barrier that was still mechanically strong? By the end of 1942 everyone thought that such a barrier did not exist. The nickel barrier had shown some early promise, but was too brittle and fragile. A barrier made of nickel powder provide to have poor separative qualities, but appeared the more likely to be successful. The experts thought there were so many potential variations and combinations that they ought to find one that worked. During experiments in 1942 they had seen that even a minor change altered considerably the separative qualities of the barriers. However with so many options they tested hundreds of different barriers, but always felt that there were more that could be tested. At the end of 1942 the ‘Norris-Adler’ type family of barriers looked the most promising.
The approach with gaseous-diffusion has been likened to a cook-book. Was the difference between a good and bad barrier just a question of personal touch? Being empirical perhaps it would all depend on some intuitive innovation, Did one sample represent a type of barrier as a whole? If they found a good barrier could they work out how to reproduce it? If they found a good barrier, could they make a lot of it?
There was progress, the ‘Norris-Adler’ barrier had been limited to laboratory trials in 1942, but in January 1943 it was decided to try and develop a continuous production process for the barrier, and in July it was operational in the laboratory in Columbia University. By that time the construction of the full-scale barrier production plant had also been started, and the company contracted to do the work (Houdaille-Hershey) also built its own pilot plant. In mid-1934 the results were not totally negative, and the plant started to produce barriers for testing. The material was susceptible to corrosion and plugging, but the biggest problem was the lack of uniformity, and in particular the separative quality. Barriers from the same batch would be totally different, so good, others not. The approach of Columbia was just continue to test samples and hope.
Building and testing gaseous-diffusion test units continued during 1943, but the idea to build a pilot plant had been abandoned, even if the building of a 10-stage unit was not immediately stopped, it just languished. The best they could manage were converters about 10 cm in diameter, and in the winter of 1943 they managed to work steadily on testing small barrier samples.
Electro-Magnetic Separation
Between 1927 and 1931 Ernest Lawrence invented the cyclotron. He had certainly read about the concept of the linear accelerator put forward by Gustaf Ising in 1924, and its first realisation by Rolf Widerøe in 1928. Lawrence was interested in using the idea to study the interaction between protons and the nucleus. Theory dictated that he would need 1 MeV protons to do that, but that would have required a prohibitively long tube. Like many great ideas, it is all about transposing one idea into another context. Lawrence realised that he could use a magnetic field to confine charged particles to a circular trajectory. The trajectory in a given magnetic field would depend upon the particles velocity. He managed to build a way to apply successive resonant accelerations through the application of a radio-frequency voltage across the gaps of magnetic pole pieces, the so-called ‘dees’. In 1929 he managed to generate a 1.3 keV proton, then it was 80 keV, and in 1931 he had his 1 MeV protons.
By 1941 the cyclotron of Lawrence had become a powerful tool for nuclear physicists, but now he had a new idea. He decided to turn his old 37-inch cyclotron into a mass spectrograph. A cyclotron accelerates charged particles from a central source along a spiral path created by a static magnetic field (the ‘dees’). The particles are accelerated along that path by a varying radio-frequency electric field. Below we can see the basic components. In the photograph we can see the ‘dees’ marked with D, and 1, 2, and 3 are radial probes that can be inserted into the charged particle stream.
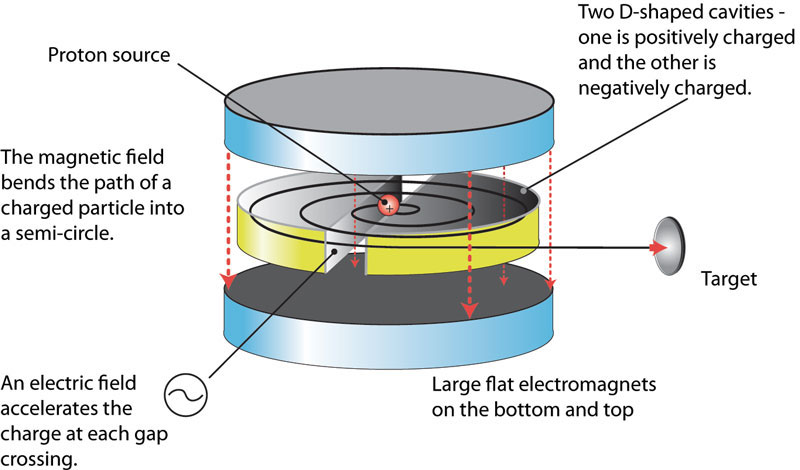

A mass spectrometer is a device that also has a ion source (e.g. one that creates ions by first vaporising and then ionising a sample), an accelerator section, an ‘analyser’ or deflector consisting of electric and magnetic fields that alters the ions trajectories dependent upon their relative mass-to-charge ratio, and one or more collectors or detectors.
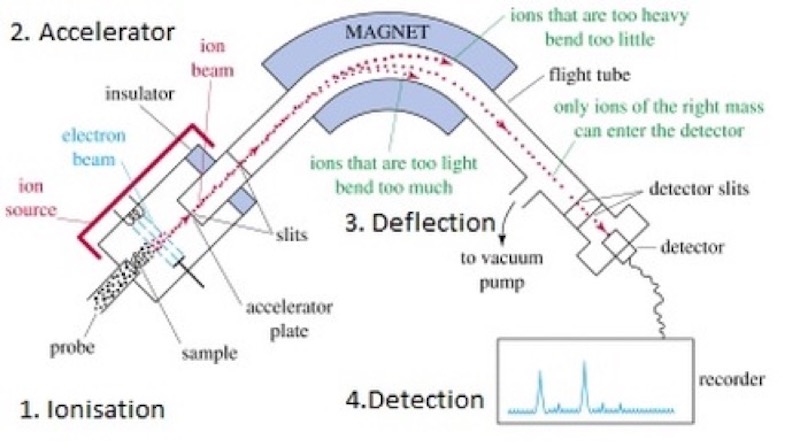
The mass spectrometer and the cyclotron have some striking similarities. Both required a high-vacuum chamber and a large electro-magnet. Lawrence knew that Nier in Minneapolis could not produce enough uranium-235 samples, so his idea was to try to obtain larger samples of uranium-235. It is true that the mass spectrometer of Nier did separate uranium-235 from uranium-238 but the quantities were extremely small (it would have taken 27,000 years for his single mass spectrometer to separate one gram of uranium-235).
The idea of electro-magnetic separation must have been in the back of Lawrence’s mind, but he also knew that there was a problem. It was generally believed that with a larger beam of positively charged ions, they would start to repel each other in the same way electrical charges do (this is called the space-charge limitation). A good example is if you imagine a current flowing through a vacuum between a cathode and an anode. That current can’t exceed a certain maximum value because of the modification of the electric field near the cathode. In simple terms the positive ions don’t see the negative cathode because the other positive ions in front of them are shielding it. So the assumption was that if there were too many positive ions they would repel each other and the scattering would disrupt the separating effect. All the texts I’ve read state that from his experience with the cyclotron Lawrence thought that the air molecules in the vacuum chamber would have a neutralising effect, i.e. negatively charged particles in the air kept the beam from dispersing under its own influence.
Starting in late November 1941 Lawrence replaced the cylindrical vacuum tank that occupied the 8-inch gap between the magnet poles of the cyclotron with the components of a mass spectrometer (similar to the one used by Nier). In the first part of the source electrical heaters vaporised solid uranium chloride. In the second part of the source electrons from a heated cathode ionised the gas. A slit 2-inches long and 0.04-inches wide permitted the positive charged ions to escape into the vacuum tank. In a plane perpendicular to the magnetic field, the beam followed a 180° circular path about 60 cm in diameter to a detector on the opposite side of the vacuum tank. The lighter uranium-235 ions would be bent just a little more, and a moveable shield with a very narrow slit enabled the operator to select which part of the beam should be collected. The tests worked well and they managed to demonstrate an enrichment to about 3%, but the quantities were of course micro-grams. The key message was that even with this device the beam current was about 5 micro-amperes, or 10 times more than that Nier’s mass spectrometer. And the way it operated showed that the space charge did not affect the beam. Within a month they had increased the beam current to 1.4 milli-amperes, and had produced three 75 micro-gram samples of 30% enriched uranium.
The objective of this device was different from a mass spectrometer. In an ionised gas containing two different isotopes, in any given time the lighter ions will travel a greater distance than the heavier ions. High frequency electrodes placed along the ion path could be used to trap one isotope and let the other continue to the target (e.g. a kind of linear chopper). This way there was no need to focus the beam on a slit, and all the ions could move in all directions. And there would be no need to bend the ions in a precise circular path. In fact there were a number of different ways to separate and collect ions from different isotopes, and the bigger the weight difference, the better they worked. The problem was to develop a technology to produce large quantities of uranium-235 quickly.
By February 1942 Lawrence had already planned a better model exploiting the 37-inch magnet. He replaced the cyclotron tank with a C-shaped vacuum chamber (rather than a large flat disk).
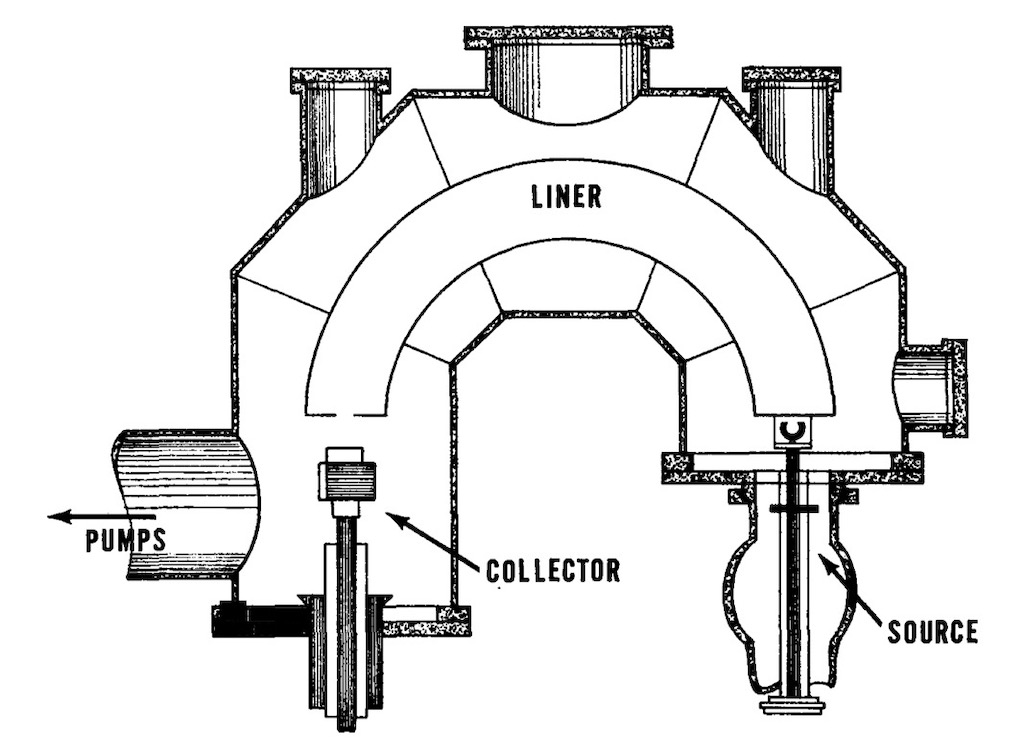
A new 10 milli-ampere source was developed. Two narrow slits focussed the beam, and the ‘unwanted’ ion’s were captured in the box structure. The intensity of the captured beams required a water-cooled collector. A water-cooled copper liner inside the tank between the source and collector was insulated so it could be maintained at the same high negative voltage as the accelerating electrode, thus keeping the ion path free from parasite electric fields that might distort the beam. This new unit was called a ‘Calutron‘. I’ve read that the name came from CalU for the University of California, and ‘tron’ a Greek suffix meaning instrument.
The results of Lawrence and the fact that new estimates of the critical mass (closer to the lower end of the range 2 kg to 100 kg) suggested that they might be able to shave 6 months of the time to complete a bomb. Oppenheimer now estimated a critical mass of 2.5 kg to 5 kg, instead of his original 100 kg. He also estimated a yield of 6% rather than the original 2% (yield in the sense of mass converted into energy).
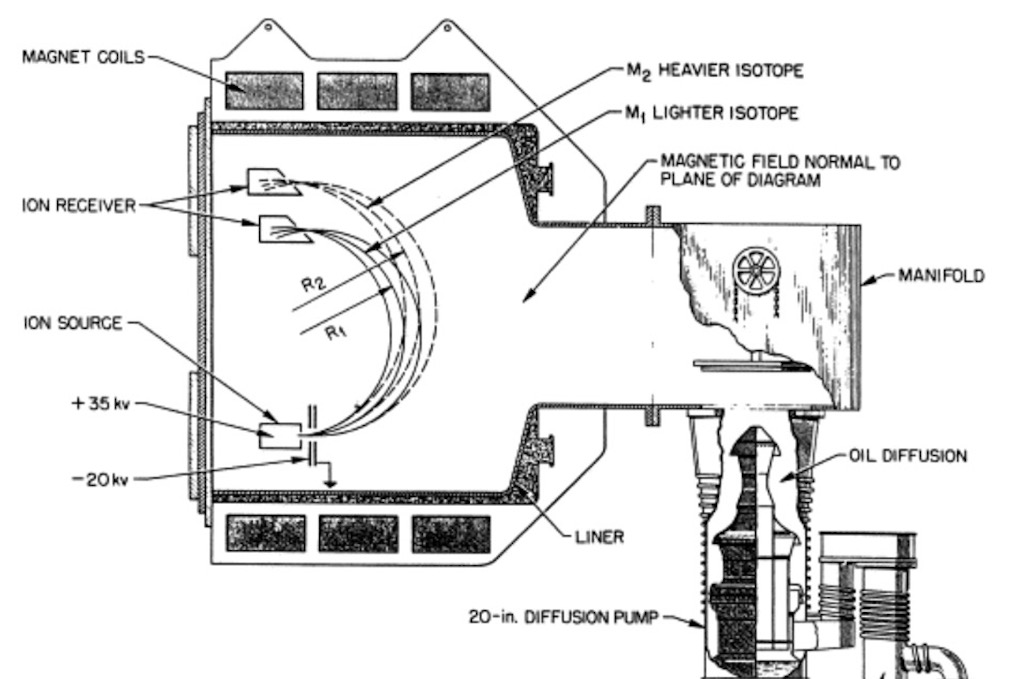
Let’s look in a bit more detail how a calutron works (above we had the so-called Beta-calutron but it might help understand better how the machine works). We have ion’s passing between the poles of a magnet. A mono-energetic beam of ions of naturally occurring uranium will split into several streams according to their momentum, one per isotope, and each characterised by a particular radius of curvature in the magnetic field. The diameter of an ion’s trajectory depends on the strength of the magnetic field, and the mass, electric charge, and speed of the ions. Lighter ions are more easily deflected by a magnetic field, and consequently have a tighter trajectory (smaller diameter). Between uranium-235 and uranium-238 the diameter of the 235U+ion beam is about 0.6% smaller than that of the 238U+ ion beam. In an electro-magnetic isotope separator with a beam trajectory diameter of 3-4 m, the difference in beam trajectory diameters for the two ions is about 0.7-0.8 cm. To properly focus these beams on their respective collector slits the voltages for ion acceleration (30-35,000 eV) and the current for the magnets must be controlled to within 0.1%.
There were many practical problems. They needed to avoid sparking in the ion source. They needed to figure out how to extract the uranium-235. Some documents mention that they finally decided on using graphite collectors that were disposable and could be burned during the chemical recovery of the uranium. The same reports also mention the need to remove the valuable feed material that had condensed (with the chloride) all over the inside of the tank (over 90% of the feed was deposited on the liners). Some reports mention that the solution was to use disposable stainless steel, water-cooled liners. However there is ample evidence indicating that the source, targets and liners were removed in one piece, and that the liners were hand scraped and brushed over larger sinks. After the cleaning the liners, sources and collectors were leak tested and reused. They also needed to scrape clean the slits to keep the beam intensity constant.
We mentioned the issue of space-charge. The technique used was to reduce the vacuum so that the positive ion beams collided with gas molecules in the vacuum chamber, creating electrons that tended to neutralise the repulsion within the beams. This was a delicate adjustment because relaxing the vacuum also reduced the beam intensity. On the 184-inch calutron the magnets were typically nearly 2 metre in diameter and weighted between 10-20 tons, each contained about 400 metres of thick copper wire. These magnets consumed about half of the energy consumption of a calutron, and naturally they also needed to be cooled.
Above we have used one of the calutron production units to describe the basic functioning of the isotope separation process, however the transition from the test unit in the 37-inch cyclotron magnet to the industrial-scale beta-calutron was not an easy one.
The test with the 37-inch magnet was sufficiently interesting for Lawrence to look to test a similar system in the 184-inch magnet. On June 3, 1942 they installed and tested two C-shaped flat vacuum tanks inside the large magnet (see below).
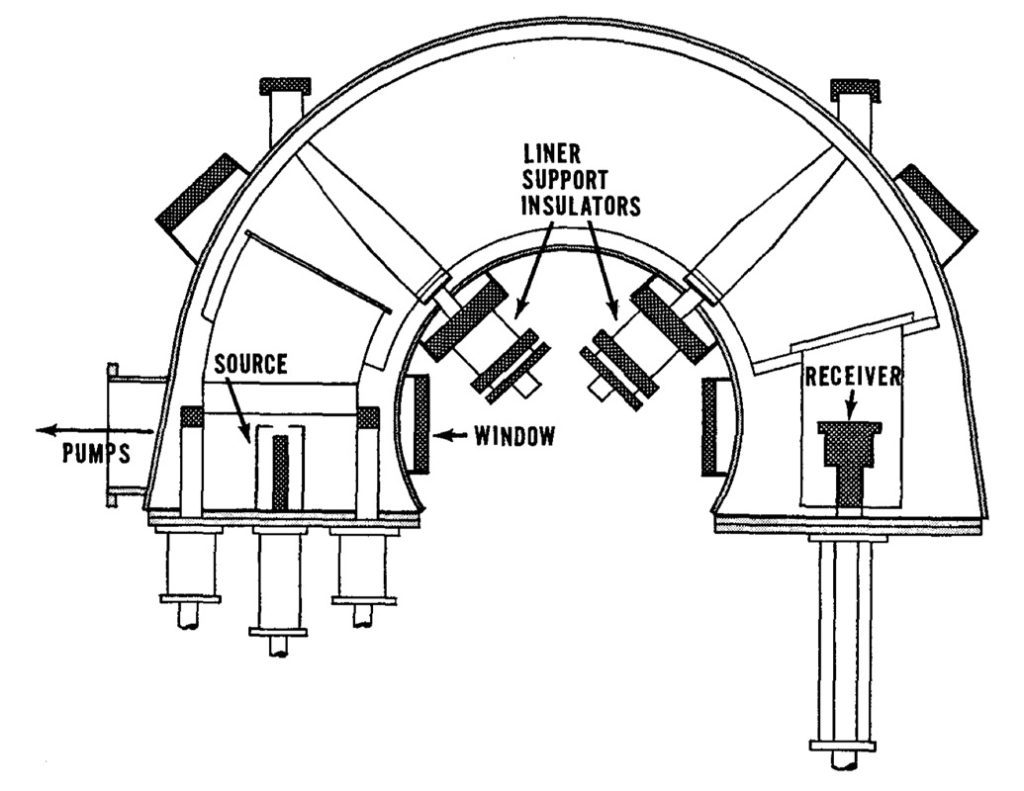
The operator would sit in front of the C-shape, with controls on the left to adjust the ion source. The operator could control the temperature of the heaters which vaporised the uranium chloride, the temperature of the charge chamber, and the power supply to the cathode which ionised the vapour. They could change the position of the cathode or replace it through an air-lock. The operator could also move the arc and the accelerator electrodes in all directions, tilt them, and change the width of the gaps. There was a vacuum-tight window through which the operator could see the adjustments made and the effect on the beam. On the right side were the controls for the collectors. Between the two was the vacuum tank, and an insulated metal liner which could support a very large negative charge.
The above calutron was only tested once for production of uranium-235, the real interest was to understand how to use at the same time several sources in one tank. The trial was with a three-beam unit, each beam about 3 cm apart from the others. It did not produce a definable beam. Nor did trials with just two sources. Single sources worked fine, and two source run at low power could operate together. What was happening was that each source generated a very high frequency oscillation in the other beams. The idea of multiple sources would bring a practical electro-magnetic plant nearer, but understanding the problem did not mean solving the problem. The other tank was used for running focusing experiments and testing different receiver designs.
Building a device that works is one problem, but building an ‘industrial’ version is another problem. Focus had not been an issue for a test unit, but the reality is that no matter how small the slit was the ions never emerged in exactly parallel paths. So the beam was blurred at the receiver. To solve this, one option is to make slight variations in the magnetic field. This was done by carefully designed contoured metal sheets or ‘shims‘ attached to the top and bottom of the vacuum tanks.
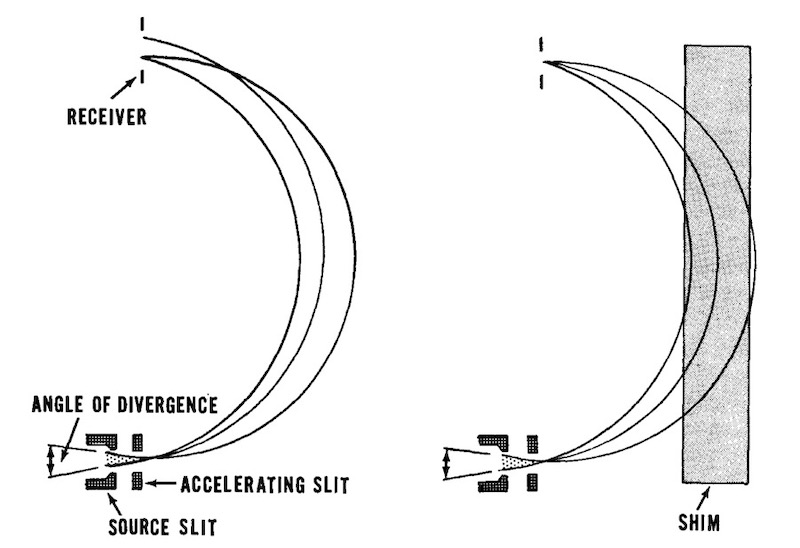
These shims varied very slightly the width of the magnet gap across the pole face. This changed the strength of the magnetic field so ‘pulling’ the ions in the beam back to focus. It literally took months of trial and error to find the best compromise. The collectors were in fact detectors, and needed to be re-designed so that the uranium-235 could be recovered. The ion beam was intense and could easily burn away a metal or graphite collector. The more intense the beam the more ions would ‘bounce out’ of the collector. Using multiple sources would need multiple collectors, but there was a problem of beam interaction. The use of shims could be optimised for one ion beam but might adversely affect another ion beam. A new problem emerged and that was the beam of ions no longer struck the face of the collector in a straight line, but as a curve. But this then required slots that were curved. And this was all about trying a design, photographing its effect through a window in the tank, and reshaping the slots for another test. This might have been tiresome but it also revealed a new result. What they found was that the collection was improved when the face of the collector was set at 45° rather than perpendicular to the beam.
The result of all this experimentation was that they knew how to produce a tight beam and they could operate two beams within a few inches of each other. For Lawrence the theory was solid, so the remaining problems were ‘just’ engineering and the chemical extraction of the uranium-235.
Finally when Lawrence had tested fully the calutron he suggested building 10 of them, to produce 4 grams of uranium-235 daily. The S-1 Committee over-ruled him and recommended spending immediately $12 million to build a plant for 25 times that capacity to be operational before autumn 1943 (a different report mentions a five tank pilot and a full-size plant with 200 tanks). Everyone knew that the alternatives would be better, but in mid-1942 the calutron was the only thing that worked. From end 1943 50 new calutrons were installed monthly, and by the time the war ended Y-12 would house 1,152 calutrons operated by hundreds of women working shifts 24/7. The women operators had to watch the measured beam currents and make minimal adjustments to operating voltages and tank pressures. Women did the job properly, whilst men tended to constantly fiddle with the controls.
Twenty-five years after the end of the war there was an anniversary meeting held at Oak Ridge. General Leslie R. Groves noted that at the time no one thought that electro-magnetic separation would work, and the only reason they continued was because it was backed by Lawrence. And he delivered, even if later gaseous-diffusion delivered larger quantities of uranium-235 and the Y-12 plant was closed down. Groves note that they had in fact kept working two alpha and two beta calutrons for isotope separation and research, and they had delivered over that 25 years more than 200 kg of enriched isotopes for science and medicine.
Isotope Separation - who decided what, and how?
Later we will see some of the major decisions made concerning isotopic separation, within the context of the different plants built in Oak Ridge. Below is a kind of experiment, in that I’ve tried to tie those decisions together in a rough time-line so we can see better the ‘play’ of one isotope separation technique against another, and how they all came together in the end.
After a meeting on the March 16, 1939 between Pegram of Columbia and the Navy, they contacted Beams about using a centrifuge for isotope separation. It was on May 21, 1940 that Kistiakowsky suggested gaseous-diffusion as a possible means of isotope separation. It was in spring 1941 that Urey took command of research on gaseous-diffusion. It was in 1941 that it was suggested that the electro-magnetic separation method could yield a few grams of uranium-235.
During the spring and summer of 1940 it was gas thermal-diffusion that first attracted the attention of scientists. The idea is simple, molecules in a mixed gas will concentrate in either the hot or cold regions of a container. But the specialists decided that it was impractical for large-scale separation. It was Philip Hauge Abelson who thought to try liquid rather than a gas. He received continued funding by the Naval Research Laboratory.
By April 1941 progress on the centrifuge appeared to be good. Tests in principle were positive, and the first trials with gaseous uranium compounds achieved a degree of concentration of uranium-235. A body of theory was established by Karl Paley Cohen and a 36-inch design was developed. In July 1941 $95,000 was allocated to the centrifuge, $25,000 to gaseous-diffusion, and $167.000 for chain reaction work.
It was in July 1941 that Lawrence decided to convert the 37-inch cyclotron to a giant mass spectrometer, and the calutron and electro-magnetic separation was born (even if Lawrence’s initial plans was just for sample preparation).
On November 6, 1941 a report by the National Academy of Science concluded that gaseous-diffusion looked feasible, even if it might be slow to develop. The centrifuge method appeared practical and was further advanced. They also stated that it might take three or four years, and the separation process would be the most time-consuming and expensive part. They estimated that it would cost $50 million to $100 million, plus $30 million to produce the bombs. The report did not mention using element-94.
However in December 1941, the centrifuge had become the second best approach to isotope separation, overtaken by gaseous-diffusion (even if it would be more expensive and may take more time to build a plant). The MAUD report in mid-1941 had clearly sided with gaseous-diffusion for isotope separation. Some reports mention an allocation of $400,000 for Lawrence and electro-magnetic separation, largely based upon the very positive early results with the modified 37-inch cyclotron.
In December 1941 during a re-focusing of the research, Urey took charge of separation by both the gaseous-diffusion and the centrifuge methods, as well as work on heavy-water. In early 1942 experiments with the different separation techniques showed that they worked for lighter elements where the relative difference in weights was large. However no one method seemed to have the capability of the mass spectrometer (electro-magnetic separation) to produce large quantities of uranium-235 in the short time available.
The reality as one author put it was that the “conclusions … were based upon great masses of postulates held together by a thin thread of experimental fact“. In fact decisions were not being made on new knowledge, but simply on a greater confidence in the hastily drawn conclusions made in late 1941.
In January 1942 it was decided to build at least one experimental centrifuge and one gaseous-diffusion unit of industrial size. The also decided to design pilot plants using a number of separative units to demonstrate feasibility. Finally they also need to work to secure adequate supplies of uranium oxide, metal and hexafluoride.
Columbia University already had a development contract for centrifuges with Westinghouse, so they were asked to obtain the parts for 24 36-inch units for a pilot plant. The Westinghouse units were scaled up versions of a 7.2-inch laboratory model. Assuming a working centrifuge, a plant producing 1 kg of very pure uranium-235 would need between 40,000 and 50,000 36-inch centrifuges. Standard Oil Development Company was contracted to study the building of a full-scale plant. The Columbia group were also looking to a 132-inch centrifuge and an initial plant of 624 123-inch centrifuges in a 15-stage cascade. But for a production of 1 kg/day they would still need about 10,000 of these large centrifuges. An alternative was to focus on 100 gm/day which would ‘only’ require about 6,000 of the 36-inch centrifuges. Westinghouse said that once they had the order, they could make 1,000 centrifuges a month. In late February 1942 Westinghouse was asked to do the engineering study for a 100 gm/day plant, a decision that had little significance give the problem encountered during the winter of 1942.
The reality was that in early 1942 everyone was beginning to understand that the engineering development of both gaseous-diffusion and the centrifuge would take time.
In early 1942 Beams had completed two runs with uranium hexafluoride through the 36-inch simple flow-through centrifuge. In theory it should have managed an enrichment from 0.72% to 0.73%, but the different was too small to measure. Westinghouse was having to focus on just designing a single industrial scale centrifuge, never mind a pilot plant.
In early 1942 the key problem was developing a metal barrier for gaseous-diffusion. This was such a complex issue that they could not even decide on how to build a large-scale barrier production plant.
Aside from the specifics of each separation process, all of them required large quantities of uranium hexafluoride. Until 1940 only gram quantities were produced, but in 1940 Philip Abelson working at the Naval Research Laboratory found that he could easily and safely produce uranium hexafluoride by fluorinating uranium tetrafluoride rather the metallic uranium. By the spring 1942 a pilot plant was operating, and a rapidly expanding production was predicted.
By May 23, 1942 neither team had produced a single operating centrifuge or gaseous-diffusion unit of any practice size. No ‘short-cuts’ had been found, and there was no way to ‘pick-a-winner’. On the other hand Lawrence was expecting to have firm evidence on electro-magnetic separation (calutron), enough to justify a full-scale plant. No one was able to draw any conclusions about the relative merits of gaseous-diffusion and the centrifuge. Maybe this would change once pilot plants were built and tested, but that would take another 6 months. And no one needed a plant making 100 gm/day. They needed to design and build a 1 kg/day plant. They finally decided to build a 100 gm/day centrifuge plant for January 1944, at a cost of $38 million (it was never built). They decided to build a pilot plant for gaseous-diffusion and look at the engineering for a production plant. And they decided to build a 100 gm/day electro-magnetic plant to be completed by September 1943 at a cost of $12 million. Added to $25 million for the piles for the production of element-94, the total budget was $80 million (plus $34 million annual operating costs). The target was a ‘few atomic bombs’ by July 1944, and about twice as many each year thereafter. Deciding all this was one thing, convincing the Army was another. They agreed to the work on the piles and electro-magnetic separation, but concerning gaseous-diffusion and the centrifuges they decided not to decide.
In July 1942 people were increasingly interested in the progress made on electro-magnetic separation, and the question if that progress justified a full-scale electro-magnetic plant. Another question was the possible feasibility of producing element-94.
During the period May-October 1942 Beam had succeeded in running a sample of uranium hexafluoride three times through his experimental centrifuge, proving that enrichment was feasible. However when the samples were analysed they were only about 60% of the predicted enrichment. Looking back this alone should have been enough to kill this option. For the 100 gm/day plant the first estimate was for 8,800 machines, but based upon the laboratory trial the number of machines needed rose to 25,000. Also the hold-up of uranium in the plant rose from 4 months to 1 year. There were two remaining options. The first was to improve the build-quality of the machines. The second was to drop the flow-through principle (used in the laboratory) in favour of a theoretically more efficient but more complex counter-current design. This technique would reduce the number of machines by 20% and the machines themselves would be smaller. Trials were started, but at the same time Westinghouse was having problems testing the 36-inch flow-though model. There were severe instabilities at critical vibration frequencies. On top of that the gas-tight, corrosion-proof seals were not working properly. There were frequent failures of the motors, shafts, and bearings at high speeds. The way Beams had designed the centrifuge it consisted of a coaxial mounting between two drive shafts. The tube was then surrounded by the critical vacuum casing, and the drive shafts protruded through openings in the casing. The protrusions were made vacuum-tight with heavy-grease seals. The ends fo the drive shafts were then supported outside the case with conventional rigid bearings. The seals and bearings resulted in a tremendous loss of energy in the form of heat. They were losing about 1 kW as compared to a 1 W for a modern centrifuge. Friction meant that these components simply wore out quickly. This type of equipment was fine for short laboratory trials, but was totally inappropriate for industry-scale operation. The 36-inch machines were delivered on May 29, 1943 and started operation in August 1943, but by September 10, 1943 there were problems. Westinghouse still continued with a pilot plant in Bayonne, New Jersey, but they needed to solve all the problems for the 24 centrifuge pilot plant. By October 1943 the experimental counter-current centrifugebuilt in Virginia had worked for only short periods, and it was difficult to see how a reliable unit would be available in the near future. Many of the engineers in the different teams were not sure that the problems could be solved and a full-scale plant built.
During a meeting on October 26, 1942 it was evident that none of the separation techniques came out on top, but the centrifuge was definitely the weakest. Gaseous-diffusion looked feasible, but would probably not be completed in time (i.e. for July 1944). Neither technique was likely to deliver before early 1945. The need for a gaseous-diffusion pilot plant that would take a year to build and test was questioned. The pilot would not have produced useful quantities of uranium-235. A full-scale plant could be completed almost as quickly as the pilot. Progress in Chicago with the pile was good, and full-scale production was planned for spring 1944. So Lawrence’s electro-magnetic technique was the best bet. It probably could deliver a kilogram by January 1944, and they could see that it could produce 100 gm/day of uranium-235 through 1944. But the requirements were just staggering to everyone, including Lawrence. Even so, Lawrence promised that there were no fundamental difficulties to build a large-scale electro-magnetic plant.
During the period September 28, 1942 to November 10, 1942 things moved quickly. It was agreed that the electro-magnetic technique would go directly to a full-scale plant, bypassing the pilot plant. They had already decided to do the same for gaseous-diffusion, so it was time to eliminate the centrifuge development. And Stone & Webster and Du Pont would develop the full-scale pile and electro-magnetic plants. Kellogg would continue on the gaseous-diffusion, with a view to a 600-stage plant.
So the priority was on the electro-magnetic technique, then gaseous-diffusion, and finally the pile (and plutonium separation). Laboratory work would continue on the centrifuge and ‘isotron‘ (a device that, unlike the calutron, used an electric field rather than a magnetic field to separate uranium isotopes).
On December 2, 1942, the Lewis Committee reported that they were impressed by the possibilities of gaseous-diffusion, but there were less impressed by the partnership Kellogg-Columbia. At the same time Compton was promising a bomb with element-94 (plutonium) in 1944 (even if at this moment Fermi had not yet demonstrated a chain reaction). The Lewis committee actually concluded that gaseous-diffusion had the best chance of success, and they recommended the design and construction of a full-scale gaseous-diffusion plant. The committee also recommended to continue work on the calutron, but with the equipment available a full-scale plant would require 22,000 calutrons, which was considered impractical. But they did recommend building a small plant with 110 calutrons in order to have uranium-235 samples for testing.
Bush sent a report to Roosevelt on December 16, 1942 concerning the bomb. The centrifuge had been eliminated, and the recommendation was to build a full-scale gaseous-diffusion plant for $150 million, plutonium plants for $100 million, and a small electro-magnetic plant for $10 million.
The key decision after December 16, 1942 was to try to transfer design responsibilities from the universities to experienced engineering companies, for example the electro-magnetic plant had been reduced to 500 tanks so that it could be built as fast as possible, but Stone & Webster could order all the material they needed for the full-sized plant (responsibility had moved from Lawrence to the Army and then to Stone & Webster as prime contractor). Groves also contracted Union Carbide to build the gaseous-diffusion plant. After this decision things started to move faster on the electro-magnetic plant. Westinghouse had to make the tanks, liners, sources and collectors, General Electric the high-voltage equipment for the magnets and tanks, and Allis-Chalmers for magnet coils. The planning was to deliver 50 tanks a month, and to have all 500 working by the end of 1943.
Oak Ridge would be home to Y-12 (electro-magnetic) plant, X-10 (plutonium-producing pile) and K-25 for gaseous-diffusion. By early 1943 the cost of this site had risen to $492 million.
In January 1943 a different approach was re-discussed. The idea was to eliminate the upper stages of the gaseous-diffusion plant (a so-called ‘squared-off’ cascade) and substitute those upper stages with either centrifuges or calutrons. This decision was finally made in August 13, 1943, and the gaseous-diffusionplant would be limited to 50% enrichment, and would feed electro-magnetic enrichment in Y-12. Given that the ground had not yet been broken on the building of K-25, it was also decided to double electro-magnetic enrichment in Y-12.
In February 1943 having seen the centrifuge option abandoned, work continued on improving the reliability of the machines. The option that the centrifuge might be used to ‘top-off’ enrichment from a gaseous-diffusionplant was kept open until September, when it was decided in favour of a ‘top-off’ with electro-magneticmachines (e.g. K-25 feeding Y-12).
During 1943 Urey had become increasingly pessimistic about gaseous-diffusion. Why had they continued to work on the Norris-Adler barrier when it had proven to be unsatisfactory? Urey was of the opinion that war projects should not rely on a major research effort, and by this time there were 700 people working on gaseous-diffusion in Columbia (and several hundred people elsewhere). Urey thought that they should not spend more money on this technique, and not build K-25. On the positive side there was some progress in make a nickel powder barrier, and the cascade design proposed by the British looked impressive. Groves had in any case decided to complete K-25. The investment already committed was just too big and Kellex (the K-25 contractor) already had 900 people working on the plant design. They had invested $5 million is a plant for the production of the barrier, and $4 million in a plant for the pumps.
Groves replaced Urey with Lauchlin M. Currie from the National Carbon Company, and Hugh Stott Taylor continued the work on the new barrier. Thus Urey was removed from the gaseous-diffusion project, one that he believed would surely fail.
If gaseous-diffusion (K-25) was to deliver in 1945, then the barrier would need to be produced by summer 1944. A meeting on November 5, 1943 offered some hope that the new nickel-powder barrier would deliver, despite the Army still planning to produce the older option. And by January 4, 1944, Union Carbide had agreed to take on the new barrier. This decision meant setting aside 2 years work on the old Norris-Adler barrier, and committing K-25 to the new barrier. One of the advantages of the new barrier was that it was simple to produce by hand, but it would need thousands of people doing simple batch work. Although it did imply ripping out the machines for producing the old barrier, and installing a new process. Summarising, they would build a fabrication facility for an untested barrier in four months, and produce all they needed in the following four months, and have K-25 running within a year.
Changing to the new barrier did not mean abandoning the older Norris-Adler barrier, it just meant that it stayed in the laboratory. In fact, with a new team, results with the older barrier started to look more promising, even if it required a much more complicated manufacturing process than the new barrier. By April 1944 it looked as if they had two different barriers made in two different laboratories. In early 1944 only about 5% of the new barriers were within the specification, but by April 1944 it was up to 45%. By June 1944 they were producing pumps, coolers, compressors and converters by the thousands. And they had about a half million valves and about 1 million metres of piping ready. Already in April 1944 six stages had been installed in K-25, all without the barriers. At that time nearly 20,000 people were working on K-25, and the cost had climbed to $281 million.
As the contractors for the electro-magnetic plant started design work they were able to introduce modification that would double the capacity of the 500 tanks. But going from the laboratory to a full-size plant also meant that changes affected other decisions that everyone thought were final. The tanks grew from 3 meters to over 4 meters in height. When the use of two sources had been accepted, Lawrence started to look at four, eight or even sixteen sources. Some of Lawrences team supported the idea of two types of calutrons, one using the enriched material from the first five ‘racetracks’. On March 17, 1943 they decided to stick with the basic calutron design, but the idea of two different types of calutron won through. The calutrons under construction were re-named ‘Alpha’ and the second stage calutron was called ‘Beta’. The five ‘Alpha’ racetracks would feed only two smaller ‘Beta’ racetracks. The two magnets would contain 36 tanks, and 72 sources. There were a lot of positives. The ‘Beta’ racetracks were smaller, could be pumped down faster, and would have shorter run times. The down side was that a single loss in a ‘Beta’ would mean losing several ‘Alpha’ runs.

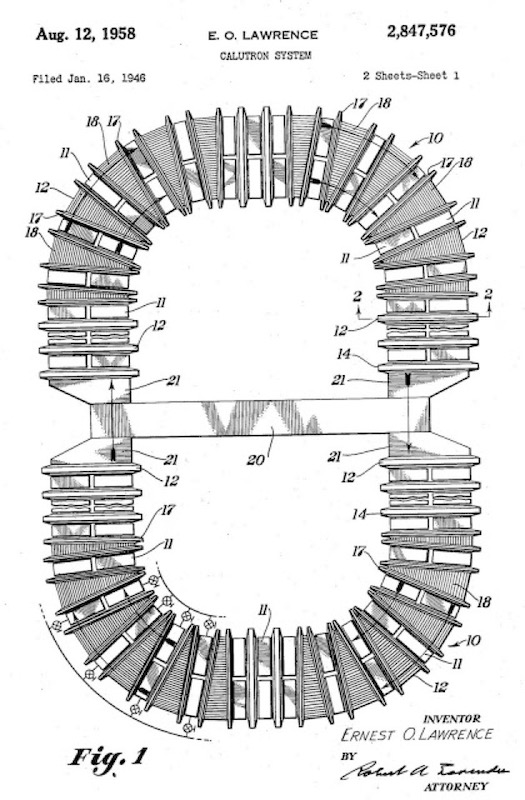
Above we can see an Alpha ‘racetrack’ with some calutron tanks sitting in the bottom left corner. And below we can see the general layout of a ‘racetrack’ taken from a patent. We see that it is a closed series of alternated tanks (no.11) and electro-magnets (no.12). The electro-magnets consist of a winding enclosed in a case (no.14) and surrounding a core which is made up of honeycombs or grids. The electro-magnets in the curved region comprise a pair of windings each is a separate winding case (no.17) with a portion of the wedge-shaped core exposed (no.18). The mid-yoke (no.20) is a beam of iron, and in order to prevent distortion of the field the end portions (no.21) are specially shaped.
One ‘Alpha’ racetrack was tested in August 1943, but it was not sure that all five racetracks would be working by the end of 1943. However, Oppenheimer emerged from Los Alamos with a three-fold increase in the amount of uranium-235 that would be need for a bomb. Would Y-12 ever produce enough material for a weapon? On the other hand the problems with the barrier for the gaseous-diffusion process meant that the electro-magnetic process was the only game in town. Maybe it could deliver enough uranium-235 for a bomb before the end of 1944. It was at this moment that the idea to eliminate the top cascades of K-25 and feed Y-12 was introduced. Naturally Lawrence used this option to argue to increase the electro-magnetic plant.
In December 1943 it was decided to let the centrifuge project run to its conclusion, but not to renew the contract. The best they had managed was to run one centrifuge for 99 days before it broke down.
Through 1944 Groves followed the idea that K-25 would feed the electro-magnetic separators in Y-12. If K-25 only provided a low enrichment, then this would feed into the ‘Alpha’ racetracks. But to do this the ‘Alpha’ machines would need different receivers. More importantly a lot more care would be needed in cleaning the ‘Alpha’ liners and recovering the now valuable charge in uranium-235 obtained from K-25. More importantly the latest design of the ‘Alpha’ racetracks had ‘simplified’ things, i.e. expecting to only work with natural uranium the liners had been removed, and only the source and receivers were to be washed. Now they would have to be completely redesigned, and the washing area doubled in size. The recovery process would need to be optimised and the risk to accumulate a critical mass in the process would need to be eliminated. Lawrence had already argued to expand the ‘Alpha’ to nine racetracks, and now he argued to add another four racetracks.

Above we can see the liners being washed, and below the collection slits from the Beta calutron for one isotope pair being cleaned and replaced.

On March 30, 1944 there was a meeting with all the major actors, and as can happen, each went away with a different opinion on what was decided. To some it looked as if gaseous-diffusion would feed Y-12 and no more electro-magnetic racetracks were needed, to others it looked as if uranium-235 had almost been dropped in favour of plutonium. Lawrence thought that K-25 might never work, so to him Y-12 was the only answer. His idea was to upgrade the existing ‘Alpha’ model to four beams and the new model to eight beams, and build 2 more ‘Alpha’ racetracks. On June 30, 1944 it was decided not to modify the existing ‘Alpha’ racetracks, but the new ‘Alpha’ racetracks would be completely redesigned with a cold source and 30 beams. Many people thought that even a combination of K-25 and Y-12 would not produce enough material for a bomb in 1944, or even early 1945.
At this point we will need to ‘back-track’ a little in time and again look at the work of Philip Abelson at the Naval Research Laboratory on liquid thermal diffusion. Firstly this is an example of what is called thermophoresis, a phenomenon where in a mixture of different particles types they will respond different to a temperature gradient. The effect was first reported on in 1856 and Charles Soret gave his name to the effect in 1879. The Chapman-Enskog theory explains that when a mix of two gases passes through a temperature gradient the heavier gas tends to concentrate at the cold end and the lighter gas at the warm end. The was used by Klaus Clusius in 1938 to separate the isotopes of neon. Abelson became interested in the use of liquid thermal diffusion to separate isotopes, and he was able to achieve a separation factor of 20% between potassium-39 and potassium-41.
Moving to uranium Abelson found that hydrolysis affected uranium salt solutions (spontaneous formation of ions), so his only option was to work with uranium hexafluoride. He received funding to continue his work, but at the time it was felt that the process would not scale to an industrial plant. Because uranium hexafluoride was not readily available Abelson devised a method to produce it in quantity. His small plant provided the samples for the work in both Columbia and Virginia, and his design was used to produce the uranium hexafluoride for the weapon program.
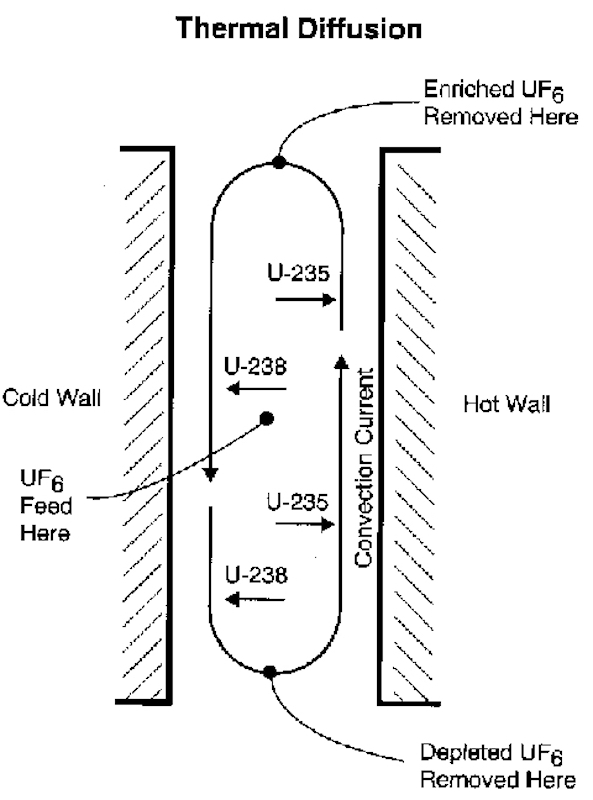
Liquid thermal diffusion is simple, uranium hexafluoride is introduced under pressure into the annular spacing between two concentric vertical pipes. The outer wall is cooled and the inner heated by high-pressure steam. The lighter isotope will concentrate near the hot wall and the heavier near the cold wall. Convection will carry the lighter isotope to the top of the column. The taller the column the greater the separation effect.
Abelson continued his work at the Naval Research Laboratory and he successfully tested the process on June 22, 1942 (a pilot plant was authorised in July 1942). Oppenheimer read the reports of Abelson, but he was convinced that it would be impossible to produce fully enriched uranium-235 by this method. By July 1944 Abelson was able to produce 5 gm/day of 5% enriched uranium. Oppenheimer now saw that the plant could actually be scaled to 100 columns operating in parallel and produce about 12 kg/day at 1% enrichment.
Early on Roosevelt had decided to exclude the Navy from the weapons development, so Abelson’s work on liquid thermal diffusion was more or less ignored. There had been some contact but the work of Abelson looked less promising (at the time) than the electro-magnetic separation and the pile for element-94. And the results obtained in Columbia and Chicago had not been given to the Navy. It was in December 1942 that they learned that Abelson had two 16 metre tall columns processing 2 kg/day of uranium hexafluoride with a significant separation. Abelson had improved the performance of his system by simply increasing the temperature difference between the two walls. The principle was very simple, had no moving parts and no valves, and was relatively inexpensive to build, but on the negative side the process took a very long time to come to equilibrium and produce enriched uranium (a large plant might take up to 600 days to reach equilibrium for fully enriched uranium). As has been noted this performance would have put it ‘on par’ with the gaseous-diffusion and centrifuge projects in 1942, but in 1943 it looked less positive.
But if liquid thermal diffusion were to be limited to 10% enrichment, then it could be considered as good as the gaseous-diffusion process planned for K-25. So why not replace gaseous-diffusion with liquid thermal diffusion? On February 10, 1943 it was decided to go ahead with preliminary engineering studies on liquid thermal diffusion, but the relationship between the Manhattan Project and the Navy would breakdown in the summer of 1943.
However Abelson continued his work, and it was in April 1944 that Oppenheimer again suggested that liquid thermal diffusion might be of value. The key to the process of Abelson was to increase the temperature difference between the hot and cold walls. In November 1943 he obtained the agreement of the Navy to build a 300 column plant in the Naval Boiler and Turbine Laboratory in Philadelphia, and by June 1944 he had build a 100 column test plant. In early 1944 it was agreed that the Army and Navy could (again) cooperate. More or less immediately Groves signed an agreement to build 21 exact copies of the Abelson 100 column test plant. Despite the fact that there were still problems with the barrier for the K-25 gaseous-diffusion plant, they had built in Oak Ridge a power plant that was ready to produce the necessary steam for the liquid thermal diffusion columns. The entire plant could be up and running within 90 days (starting June 27, 1944). This new plant was called S-50.
Making 100 16-metre tall columns was one thing, making 2,000 of them was another. The inner pipe used for the liquid thermal diffusion column was in nickel and the outer in copper, and the gap was only 0.005 cm. Within 69 day they had 320 columns working, whereas the K-25 gaseous-diffusion plant was not yet finished, and they had received just two complete converters of the 1,000’s expected, and tests were still being performed on dummy barriers.
By March 1945 S-50 and Y-12 were working. All 21 racks in liquid thermal diffusion plant (S-50) were working and feeding the electro-magnetic plant (Y-12). The precise operating characteristics of the gaseous-diffusion plant (K-25) were not yet known. S-50 was enriching from 0.71% to 0.89%. This was fed to the Alpha racetracks in Y-12, then to the Beta racetracks. When K-25 came on line the feed from S-50 would go to K-25 to be enriched to 1.1% before being sent to Y-12. Over time K-25 would be brought up to 20%, and the Alpha I calutrons in Y-12 would be phased out.
In April 1945 the Alpha II racetracks were working on 1.1% material obtained from S-50 and K-25. It was finally decided to stop shipping from S-50 to Y-12, and ship from S-50 to K-25. Finally the gaseous-diffusion plant was working. By June 1945 K-25 was supplying 7% enriched material directly to the Beta racetracks in Y-12.
Not strictly in chronological order, but throughout 1944 Groves was under pressure to commit to a long-range construction plan. He finally decided to build a second gaseous-diffusion plant (K-27) and to add a fourth Beta electro-magnetic racetrack to Y-12. Both were planned for operation in 1946. At that time Groves assumption was that the war with Japan would not be over before mid-1946.
By August 1945 K-25 was fully operational, and by January 1946 K-27 was operational. What became evident was that K-25 and K-27 were now able to obtain the same enrichments as the Beta racetracks in Y-12. The problem of a potential holdup of a critical mass in K-25 was now a problem, and another problem was the speed with which corrosion was spreading in the plant was an unknown.
The above section has tried to tie together the different developments concerning isotope enrichment and separation, but now we must return to our chronological view of events, and we do so on the day the U.S. entered WW II.